 Vector took a moment this morning at the Boston Children’s Hospital Global Pediatric Innovation Summit + Awards to catch up with the Gene Discovery Core at the Manton Center for Orphan Disease Research. Its exhibition table doesn’t have fancy mannequins or flashy screens, but this team is rocking genetics and genomics, one patient at a time.
Vector took a moment this morning at the Boston Children’s Hospital Global Pediatric Innovation Summit + Awards to catch up with the Gene Discovery Core at the Manton Center for Orphan Disease Research. Its exhibition table doesn’t have fancy mannequins or flashy screens, but this team is rocking genetics and genomics, one patient at a time.
The usual methods for finding disease-causing genes don’t work for many patients who walk in the doors of Boston Children’s, or who mail in samples from all over the world. They may be one of just a handful of patients in the world with their condition—which may not even have a name yet. Full story »
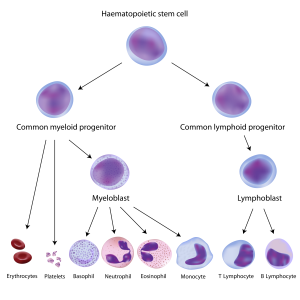
Blood-forming hematopoietic stem cells (top) give rise to all blood and immune cell types. In children with SCID, the steps leading to immune cells are broken.
In the world of fatal congenital immunodeficiency diseases, good news is always welcome, because most patients die before their first birthday if not treated. Babies with severe combined immunodeficiency disease, aka SCID or the “bubble boy disease,” now have more hope for survival thanks to two pieces of good news.
Transplants are looking up
First came a July paper in the New England Journal of Medicine (NEJM) by the Primary Immune Deficiency Treatment Consortium. This North American collaborative analyzed a decade’s worth of outcomes of hematopoietic stem cell transplant (HSCT), currently the only standard treatment option for SCID that has a chance of providing a permanent cure. Full story »
Understanding the genetic causes of nephrotic syndrome could lead to better drug treatments that reduce the need for dialysis or a kidney transplant. (Image: Wikimedia Commons)
Nephrotic syndrome is one of the worst diseases a child can have. It strikes the filtering units of the kidney, structures known as glomeruli. There’s no good treatment: Steroids are the main therapy used, but 20 percent of cases are steroid-resistant. In the syndrome’s most severe form,
focal segmental glomerulosclerosis (FSGS), children are forced onto chronic dialysis and often require a kidney transplant—often only to have their disease recur in the new organ.
Until recently, no one knew what caused nephrotic syndrome; the first causative gene was identified just a dozen years ago. The lab of Friedhelm Hildebrandt, MD, PhD, at Boston Children’s Hospital is one of a handful that’s been chipping away at the others.
Hildebrandt receives, on average, one blood sample a day from patients all over the world. Full story »

New methods can find mutations that strike just 1 in 10 cells in a sample.
It’s become clear that our DNA is far from identical from cell to cell and that disease-causing mutations can happen in some of our cells and not others,
arising at some point after we’re conceived. These so-called somatic mutations—affecting just a percentage of cells—are subtle and easy to overlook, even with next-generation genomic sequencing. And they could be more important in neurologic and psychiatric disorders than we thought.
“There are two kinds of somatic mutations that get missed,” says Christopher Walsh, MD, PhD, chief of Genetics and Genomics at Boston Children’s Hospital. “One is mutations that are limited to specific tissues: If we do a blood test, but the mutation is only in the brain, we won’t find it. Other mutations may be in all tissues but in only a fraction of the cells—a mosaic pattern. These could be detectable through a blood test in the clinic but aren’t common enough to be easily detectable.”
That’s where deep sequencing comes in. Reporting last month in The New England Journal of Medicine, Walsh and postdoctoral fellow Saumya Jamuar, MD, used the technique in 158 patients with brain malformations of unknown genetic cause, some from Walsh’s clinic, who had symptoms such as seizures, intellectual disability and speech and language impairments. Full story »

Emmie Mendes was lucky enough to be diagnosed before age 3, but many families face a much longer journey.
At first, Corrie and Adam Mendes thought their daughter Emmie had an inner ear problem. She was late with several early milestones, including walking, and when she did walk, she often lost her balance. The family pediatrician sent them to a neurologist, who ordered a brain MRI and diagnosed her with pachygyria, a rare condition in which the brain is smoother than normal, lacking its usual number of folds.
Additionally, Emmie’s ventricles, the fluid-filled cushions around the brain, looked enlarged, so the neurologist recommended brain surgery to install a shunt to drain off fluid. He advised Corrie and Adam that Emmie’s life expectancy would be greatly reduced.
As Corrie recounts on her blog, Emmie’s Story, she went online and came across the research laboratory of Christopher Walsh, MD, PhD, at Boston Children’s Hospital. The lab does research on brain malformations and has an affiliated Brain Development and Genetics Clinic that can provide medical care.
After Walsh’s team reviewed Emmie’s MRI scan, genetic counselor Brenda Barry invited the family up from Florida. Full story »
Giving patients the right kind of immune cells could curb their IBD, research suggests.
Inflammatory bowel disease (IBD) is miserable for anyone, but when it strikes a child under age 5, it’s much more severe, usually causing bloody diarrhea, wrenching abdominal pain and stunted growth. Early-onset IBD is rare, but on the rise: For reasons unknown, its incidence is increasing by about 5 percent per year in some parts of the world.
A recently identified form of early-onset IBD shows up within months of birth, causing severe inflammation in the large intestine and abscesses around the anus. Recently linked to genetic mutations in the cellular receptor for a signaling protein, interleukin-10 (IL-10), it can also lead to lymphoma later in life.
As with all early-onset IBD, IL-10-receptor deficiency has no good treatment. A bone marrow transplant is actually curative, but carries many risks, especially in infants.
“We’ve been trying to understand why IBD in these children is so severe and presents so early,” says Dror Shouval, MD, a pediatric gastroenterologist at Boston Children’s Hospital and a fellow in the lab of Scott Snapper, MD, PhD. The beginnings of such an understanding—detailed recently in the journal Immunity—could lead to a new treatment approach for this and perhaps other kinds of early-onset IBD. Full story »

Emir Seyrek was the first patient with Wiskott-Aldrich syndrome to be treated in the U.S. in an international gene therapy trial.
Seeing that his mother, Kadriye, wasn’t looking, Emir Seyrek got an impish grin on his face, the kind only a two-year-old can have. He quietly dumped his bowl of dry cereal out on his bed and, with another quick look towards his mother, proceeded to pulverize the flakes to dust with his toy truck. The rest of the room burst out laughing while his mother scolded him. Despite the scolding, though, the impish grin remained.
It was hard to believe that he arrived from Turkey six months earlier fighting a host of bacterial and viral infections. Emir was born with Wiskott-Aldrich syndrome (WAS), a genetic immunodeficiency that left him with a defective immune system. He was here because he was the first patient—of two so far—to take part in an international trial of a new gene therapy treatment for WAS at Dana-Farber/Boston Children’s Cancer and Blood Disorders Center. And that day he was having his final checkup at Boston Children’s Hospital’s Clinical and Translational Study Unit before going home. Full story »

Rather than a single drug, cocktail of approaches is most likely to successfully preserve muscle.
It’s been 28 years since a missing dystrophin protein was found to be the cause of Duchenne muscular dystrophy (DMD), a disease affecting mostly boys in which muscle progressively deteriorates. Dystrophin helps maintain the structure of muscle cells; without it, muscles weaken and suffer progressive damage, forcing boys into wheelchairs and onto respirators.
Today, a variety of approaches that attempt to either restore dystrophin or compensate for its loss are in the therapeutic pipeline.
“We’re at the point where lots of things are going into clinical trials,” says Louis Kunkel, PhD, who is credited with identifying dystrophin in 1987. “I call it the decade of therapy.” Full story »
 It was the variability that intrigued pediatric cardiologist William Pu, MD, about his patient with heart failure. The boy suffered from a rare genetic mitochondrial disorder called Barth syndrome. While he ultimately needed a heart transplant, his heart function seemed to vary day-to-day, consistent with reports in the medical literature.
It was the variability that intrigued pediatric cardiologist William Pu, MD, about his patient with heart failure. The boy suffered from a rare genetic mitochondrial disorder called Barth syndrome. While he ultimately needed a heart transplant, his heart function seemed to vary day-to-day, consistent with reports in the medical literature.
“Often patients present in infancy with severe heart failure, then in childhood it gets much better, and in the teen years, much worse,” says Pu, of the Cardiology Research Center at Boston Children’s Hospital. “This reversibility suggests that this is a disease we should really be able to fix.”
Though it needs much more testing, a potential fix may now be in sight for Barth syndrome, which has no specific treatment and also causes skeletal muscle weakness and low white-blood-cell counts. It’s taken the work of multiple labs collaborating across institutional lines. Full story »
 Perhaps counter-intuitively, rare diseases can present attractive business opportunities for pharmaceutical companies. As discussed previously on Vector, they generally offer:
Perhaps counter-intuitively, rare diseases can present attractive business opportunities for pharmaceutical companies. As discussed previously on Vector, they generally offer:
1) a population of patients with a high, unmet need, greatly lowering the bar to FDA approval
2) a closely networked disease community, greatly lowering the bar to creating disease registries and mounting clinical trials
3) well-studied disease pathways.
Recoiling from expensive failures of would-be blockbuster drugs, companies like Pfizer, Novartis, GlaxoSmithKline, Sanofi, Shire and Roche are embracing rare diseases, despite their small market sizes, because of their much clearer path to clinic. But in the current risk-averse industry environment, some projects are stalling. Industry may need more incentive to jump in—and Cydan Development is basing its business model on providing it. Full story »







