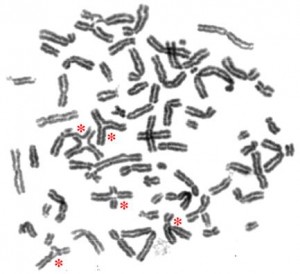
Fanconi anemia cells have multiple defects in response to DNA damage, making it hard to create iPS cells from them.
I’m attending an international pediatric oncology meeting in Boston with about 2,000 other people. In a session on gene therapy, David Williams, chief of Hematology/ Oncology at Children’s, talked about trying to treat a rare inherited bone marrow failure syndrome, Fanconi anemia, by correcting the gene in the patient’s blood stem cells. It didn’t work the first time, but he has a new tactic.
Fanconi anemia is caused by a single genetic defect in any one of 13 genes and seems to cripple DNA repair. Children are typically born with congenital abnormalities, and, at around age 8, the cells in their bone marrow begin to fail. Ultimately, the only cure is hematopoietic stem cell transplantation, which can have serious toxic side effects, especially with unrelated donors.
Williams turned to gene therapy for its potential to reverse the bone marrow failure with fewer side effects. Several years ago, he attempted gene therapy in a clinical trial for children with Fanconi anemia, starting with the few retrievable stem cells from their faltering bone marrow. The genetic correction seemed to work, but the researchers could not collect and correct sufficient cells for a therapeutic effect. They stopped the study after similar experiences with the stem cells of three patients.
Gene therapy trials in children with severe combined immunodeficiency (SCID), also known as the “bubble-boy syndrome,” appear to show that at least one million genetically corrected cells are needed to correct these diseases, said Williams (who has just begun leading a new SCID-X1 trial in the U.S). Now, the advent of induced pluripotent stem (iPS) cell technology has led to new hopes — the possibilty of creating unlimited numbers of genetically corrected blood cells.
Since iPS cells are made from a person’s own cells-typically a skin cell-the blood cells derived from them would lack many of the immunological side effects of transplanting another person’s cells. Two years ago, George Daley, Williams’s colleague and collaborator, announced the creation of 10 iPS cell lines from patients with a variety of diseases. Last month, Williams, Daley and Alan D’Andrea, another colleague at Dana Farber/Children’s Hospital Cancer Care, received a three-year, $3.9 million grant to develop gene therapy in iPS cells and other approaches to treat Fanconi anemia.
They will need to tackle a huge problem: skin cells from patients with Fanconi anemia have been notoriously difficult to transform into iPS cells. Last year, a Spanish study suggested why. The group was able to create low numbers of Fanconi iPS cells, but only if they corrected the genetic defect before reprogramming the skin cells to iPS cells. Williams and Daley suspect the Fanconi pathway itself-and the DNA damage response-is important in iPS cell reprogramming.
“It appears to be related to oxidative damage during reprogramming,” said Williams, who has taken the studies back to mice. “Healthy cells presumably take it in stride and correct any abnormality. Fanconi cells can’t handle the stress.”
The deficiencies in the Fanconi pathway decrease the efficiency of iPS cell creation and increase DNA damage. William and his colleagues restored programming efficiency nearly to normal by first restoring the Fanconi pathway and then reprogramming them in hypoxic conditions. The hypoxic environment also works to boost iPS reprogramming in uncorrected cells, which can be used to model the disease in a dish. “We can generate perfectly good hematological colonies, that when transplanted in mice can generate three types of tissues,” Williams reported.
For people, the working model begins with genetic correction of Fanconi skin cells, reprogramming them to embryonic-like iPS cells, and then differentiating them to blood stem cells.
A big unanswered question in human cells: Will the Fanconi genetic correction persist through the reprogramming process? “We would hope, but we haven’t proven it,” Williams said. That’s one of the many items on his lab’s “to do” list.






